Volume 6, Issue 1 (Journal of Clinical and Basic Research (JCBR) 2022)
jcbr 2022, 6(1): 32-36 |
Back to browse issues page


Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Hosseini S A, Rahafard S, Kia A. A Case of Late Infantile Metachromatic Leukodystrophy Presenting with Gradually-onset Paraplegia. jcbr 2022; 6 (1) :32-36
URL: http://jcbr.goums.ac.ir/article-1-353-en.html
URL: http://jcbr.goums.ac.ir/article-1-353-en.html



1- Neonatal & Children's Health Research Center , Golestan University of Medical Sciences, Gorgan , Iran
2- Neonatal & Children's Health Research Center , Golestan University of Medical Sciences, Gorgan , Iran , dr.rahafrd@gmail.com
3- Mehregan Imaging Centre , Gorgan , Iran
2- Neonatal & Children's Health Research Center , Golestan University of Medical Sciences, Gorgan , Iran , dr.rahafrd@gmail.com
3- Mehregan Imaging Centre , Gorgan , Iran
Keywords: Lysosomal storage diseases, Metachromatic leukodystrophy, Demyelinating disease, Neurodegenerative disease, Late infantile MLD
Full-Text [PDF 320 kb]
(645 Downloads)
| Abstract (HTML) (1769 Views)
Full-Text: (419 Views)
INTRODUCTION
Metachromatic leukodystrophy (MLD) is a lysosomal storage disease with an estimated prevalence of 1 per 40,000 to 160,000 around the world (1). This hereditary autosomal recessive, neurodegenerative disease affects the myelin sheath of the nerve fibers of central (CNS) and peripheral nervous systems, a pathophysiology similar to multiple sclerosis (2). Progressive alteration in motor and cognitive functions are the most common clinical presentation (2), while, muscle spasms, spastic tetraparesis, optic atrophy, and ataxia are other important clinical manifestations (3). The age of onset of MLD varies from 18 months to adulthood. The disease has a poor prognosis with a majority of cases ending up with vegetative state or death (2). Late infantile MLD is the most common form of the disease that accounts for about 50% of all cases of MLD. It usually presents with progressive decrease in visual acuity, impaired swallowing, muscle rigidity, seizures, and developmental delays (4). Juvenile variant has a worldwide prevalence of 23% and occurs at age of 4 to 12 years. It is marked as early juvenile MLD if presented before age of 6 (2, 5).
In this case report, we present a rare case of late infantile MLD in a four-and-half-year-old male patient presenting with gradually-onset Paraplegia.
CASE PRESENTATION
We hereby present a four-and-half-year-old male patient with chief complaint of
gradually-onset disability in walking. The patient had been conceived from a term pregnancy by natural vaginal delivery and had a birth Apgar score of 9 out of 10 from cousin parents. He had a normal growth and neurodevelopmental pattern until the 15th month of life. Afterward, he had a gradually increasing abnormality in gate and finally has become paralyzed since the 3rd year of life. The patient was also suffering from dysphagia, and the barium swallow test was normal. Parents reported complaint of bilateral ptosis in the patient as well as a bad temperament. The patient also had a left side undescended testis.
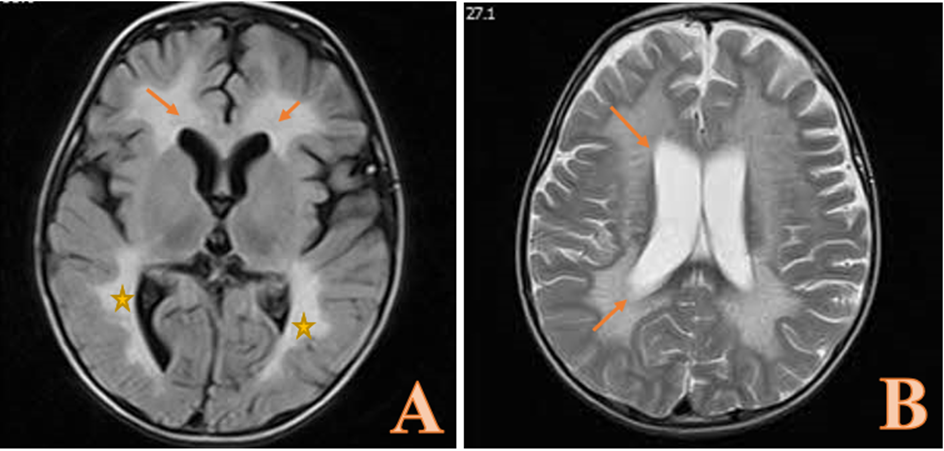
First, we assessed the patient in terms of metabolic syndromes and myopathies by measuring aldolase (7.5 units/l), creatine kinase (238 units/ l), anti-acetylcholine receptor antibodies, and anti-muscle specific kinase antibodies, which were all within the normal range. The patient underwent brain MRI without contrast (Figure 1), which indicated symmetric, high signal intensities in the parietal-occipital central white matter and frontal central white matter, with spreads to periventricular white matters in T2-weighted sequence. Upon these findings and according to the clinical manifestations, we had a high suspicion toward MLD. Whole exome sequencing revealed the possibly of pathogenic variant chr22:51064623C>G;c.938G>C(p.Arg313Pro) in exon 5 of Arylsulfatase A (ARSA) gene, which is compatible with autosomal recessive MLD. Thus, the diagnosis of late-infantile MLD was confirmed for the patient.
Metachromatic leukodystrophy (MLD) is a lysosomal storage disease with an estimated prevalence of 1 per 40,000 to 160,000 around the world (1). This hereditary autosomal recessive, neurodegenerative disease affects the myelin sheath of the nerve fibers of central (CNS) and peripheral nervous systems, a pathophysiology similar to multiple sclerosis (2). Progressive alteration in motor and cognitive functions are the most common clinical presentation (2), while, muscle spasms, spastic tetraparesis, optic atrophy, and ataxia are other important clinical manifestations (3). The age of onset of MLD varies from 18 months to adulthood. The disease has a poor prognosis with a majority of cases ending up with vegetative state or death (2). Late infantile MLD is the most common form of the disease that accounts for about 50% of all cases of MLD. It usually presents with progressive decrease in visual acuity, impaired swallowing, muscle rigidity, seizures, and developmental delays (4). Juvenile variant has a worldwide prevalence of 23% and occurs at age of 4 to 12 years. It is marked as early juvenile MLD if presented before age of 6 (2, 5).
In this case report, we present a rare case of late infantile MLD in a four-and-half-year-old male patient presenting with gradually-onset Paraplegia.
CASE PRESENTATION
We hereby present a four-and-half-year-old male patient with chief complaint of
gradually-onset disability in walking. The patient had been conceived from a term pregnancy by natural vaginal delivery and had a birth Apgar score of 9 out of 10 from cousin parents. He had a normal growth and neurodevelopmental pattern until the 15th month of life. Afterward, he had a gradually increasing abnormality in gate and finally has become paralyzed since the 3rd year of life. The patient was also suffering from dysphagia, and the barium swallow test was normal. Parents reported complaint of bilateral ptosis in the patient as well as a bad temperament. The patient also had a left side undescended testis.
First, we assessed the patient in terms of metabolic syndromes and myopathies by measuring aldolase (7.5 units/l), creatine kinase (238 units/ l), anti-acetylcholine receptor antibodies, and anti-muscle specific kinase antibodies, which were all within the normal range. The patient underwent brain MRI without contrast (Figure 1), which indicated symmetric, high signal intensities in the parietal-occipital central white matter and frontal central white matter, with spreads to periventricular white matters in T2-weighted sequence. Upon these findings and according to the clinical manifestations, we had a high suspicion toward MLD. Whole exome sequencing revealed the possibly of pathogenic variant chr22:51064623C>G;c.938G>C(p.Arg313Pro) in exon 5 of Arylsulfatase A (ARSA) gene, which is compatible with autosomal recessive MLD. Thus, the diagnosis of late-infantile MLD was confirmed for the patient.
Figure 1. T2-weighted brain MRI showing symmetric high signal intensities in the parietal-occipital central white matter () and frontal central white matter (arrows in A) with spreads to periventricular white matters (arrows in B).
DISCUSSION Metachromatic leukodystrophy is a lipid storage disorder resulting from the ARSA enzyme deficiency. A majority of patients with MLD are from Caucasian ethnicities (6, 7). Deficiency in the ARSA enzyme leads to accumulation of sulfatide (sphingolipid cerebroside 3-sulfate) in neurons of the CNS, thereby causing neuronal dysfunction and degeneration (8). Previous studies have also considered a determining role for inflammatory processes in the pathophysiology of MLD, as there have been elevated levels of interleukin-8, interleukin-1 receptor antagonist, and vascular endothelial growth factor in both plasma and cerebrospinal fluid of MLD patients (1). The disease is divided into three subtypes based on the age of onset; late infantile MLD (occurs before age of 30 months), juvenile MLD (starts at age of 30 months-16 years), and adult-onset MLD (occurs after the age of 16 years) (2). Definite diagnosis of MLD involves a complete evaluation ranging from molecular and biochemical tests to genetic and neuroradiological (MRI) evaluations (2). In early phases, MLD is usually presented with unspecific signs and symptoms including behavioral problems and focal neurological disorders, which make the MLD difficult to
diagnose (9). About 75% of cases show the initial symptoms prior to 18 months of age (10). Our patient had a gradually progressive gait problem and motor regression since age of 15th month until he got para-paralyzed at 3 years of age.
According to a nationwide cohort study in Germany, 48% of patients with late infantile MLD develop regression of speech and language, similar to our case (11). In addition, the mean time between early symptoms and diagnosis has been reported be 12 months in patients with late infantile MLD, which is similar to our case. In a case series involving 18 MLD cases (10 male and 8 female) in Iran, 80% of the patients had consanguineous parents, out of which, 45% were first cousins. In addition, 12 patients had late infantile MLD and six patients had juvenile MLD. Electromyography showed demyelinating sensorimotor neuropathy pattern in 96% of the patients (12). These results suggest that MLD should be considered in patients with ataxia, developmental regression, positive family history of MLD, and consanguinity marriage.
In another study in Iran, Golchin et al. evaluated three MLD patients from a family and identified a new mutation in the ARSA gene. They reported a homozygous missense mutation c.1070 G > T (p.Gly357Val) in exon 6 of these patients, which was reported for the first time in MLD patients. They concluded that diagnostic strategies should detect both common and rare MLD alleles (13).
Similar to our case, two studies reported periventricular white matter involvement in brain MRI of patients with late infantile MLD (14, 15). Groeschel et al. found almost no brain MRI abnormalities in late infantile MLD patients until the presentation of first clinical symptoms. Thus, it seems that early brain MRI studies may not provide enough diagnostic clues.
Currently, there are no approved treatment for MLD; however, transplantation of hematopoietic stem cells and enzyme replacement therapy are used to postpone disease progression in some patients (2).
CONCLUSION
Generally, early diagnosis of MLD is of great importance as it may increase the chance of recovery from the disease. Clinical suspicion toward MLD is the key component of early diagnosis, and adequate paraclinical tests can completely confirm the diagnosis. In conclusion, we suggest considering MLD in patients suffering from behavioral, visual, and motor regressions, especially those with normal metabolic tests. Despite absence of a definitive treatment, proper diagnosis helps physicians with better follow-up of patients and managing complications.
ACKNOWLEDGMENTS
None.
DECLARATIONS
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Ethics approvals and consent to participate
Consent was taken from parents of the patients after ensuring confidentiality of personal information.
Conflict of interest
The authors declare that there is no conflict of interest regarding publication of this article
diagnose (9). About 75% of cases show the initial symptoms prior to 18 months of age (10). Our patient had a gradually progressive gait problem and motor regression since age of 15th month until he got para-paralyzed at 3 years of age.
According to a nationwide cohort study in Germany, 48% of patients with late infantile MLD develop regression of speech and language, similar to our case (11). In addition, the mean time between early symptoms and diagnosis has been reported be 12 months in patients with late infantile MLD, which is similar to our case. In a case series involving 18 MLD cases (10 male and 8 female) in Iran, 80% of the patients had consanguineous parents, out of which, 45% were first cousins. In addition, 12 patients had late infantile MLD and six patients had juvenile MLD. Electromyography showed demyelinating sensorimotor neuropathy pattern in 96% of the patients (12). These results suggest that MLD should be considered in patients with ataxia, developmental regression, positive family history of MLD, and consanguinity marriage.
In another study in Iran, Golchin et al. evaluated three MLD patients from a family and identified a new mutation in the ARSA gene. They reported a homozygous missense mutation c.1070 G > T (p.Gly357Val) in exon 6 of these patients, which was reported for the first time in MLD patients. They concluded that diagnostic strategies should detect both common and rare MLD alleles (13).
Similar to our case, two studies reported periventricular white matter involvement in brain MRI of patients with late infantile MLD (14, 15). Groeschel et al. found almost no brain MRI abnormalities in late infantile MLD patients until the presentation of first clinical symptoms. Thus, it seems that early brain MRI studies may not provide enough diagnostic clues.
Currently, there are no approved treatment for MLD; however, transplantation of hematopoietic stem cells and enzyme replacement therapy are used to postpone disease progression in some patients (2).
CONCLUSION
Generally, early diagnosis of MLD is of great importance as it may increase the chance of recovery from the disease. Clinical suspicion toward MLD is the key component of early diagnosis, and adequate paraclinical tests can completely confirm the diagnosis. In conclusion, we suggest considering MLD in patients suffering from behavioral, visual, and motor regressions, especially those with normal metabolic tests. Despite absence of a definitive treatment, proper diagnosis helps physicians with better follow-up of patients and managing complications.
ACKNOWLEDGMENTS
None.
DECLARATIONS
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Ethics approvals and consent to participate
Consent was taken from parents of the patients after ensuring confidentiality of personal information.
Conflict of interest
The authors declare that there is no conflict of interest regarding publication of this article
Article Type: Case report |
Subject:
Medicine
References
1. Shaimardanova AA, Chulpanova DS, Solovyeva VV, Mullagulova AI, Kitaeva KV, Allegrucci C, et al. Metachromatic leukodystrophy: diagnosis, modeling, and treatment approaches. Frontiers in Medicine. 2020;7. [View at Publisher] [DOI] [PMID] [PMCID] [Google Scholar]
2. Rosenberg JB, Kaminsky SM, Aubourg P, Crystal RG, Sondhi D. Gene therapy for metachromatic leukodystrophy. Journal of neuroscience research. 2016;94(11):1169-79. [View at Publisher] [DOI] [PMID] [PMCID] [Google Scholar]
3. Mallick A, Godil A, Khetpal A, Rizvi AH, Khan F. Infantile metachromatic leukodystrophy in an 18 month old girl. JPMA The Journal of the Pakistan Medical Association. 2016;66(9):1197-200. [View at Publisher] [Google Scholar]
4. Gieselmann V, Krägeloh-Mann I. Metachromatic leukodystrophy-an update. Neuropediatrics. 2010;41(01):1-6. [View at Publisher] [DOI] [PMID] [Google Scholar]
5. van Rappard D. Mind the Metachromatic leukodystrophy: Natural evolution and treatment effects. 2018. [Google Scholar]
6. Batzios SP, Zafeiriou DI. Developing treatment options for metachromatic leukodystrophy. Molecular genetics and metabolism. 2012;105(1):56-63. [View at Publisher] [DOI] [PMID] [Google Scholar]
7. Cesani M, Lorioli L, Grossi S, Amico G, Fumagalli F, Spiga I, et al. Mutation update of ARSA and PSAP genes causing metachromatic leukodystrophy. Human mutation. 2016;37(1):16-27. [View at Publisher] [DOI] [PMID] [Google Scholar]
8. Wolf NI, Breur M, Plug B, Beerepoot S, Westerveld AS, van Rappard DF, et al. Metachromatic leukodystrophy and transplantation: Remyelination, no cross‐correction. Annals of clinical and translational neurology. 2020;7(2):169-80. [View at Publisher] [DOI] [PMID] [PMCID]
9. Mahmood A, Berry J, Wenger DA, Escolar M, Sobeih M, Raymond G, et al. Metachromatic leukodystrophy: a case of triplets with the late infantile variant and a systematic review of the literature. Journal of child neurology. 2010;25(5):572-80. [View at Publisher] [DOI] [PMID] [PMCID] [Google Scholar]
10. Borges FM, Costa MJGd, Carneiro ZA, Lourenço CM. Metachromatic leukodystrophy: pediatric presentation and the challenges of early diagnosis. Revista da Associação Médica Brasileira. 2020;66:1344-50. [View at Publisher] [DOI] [PMID] [Google Scholar]
11. Kehrer C, Groeschel S, Kustermann-Kuhn B, Bürger F, Köhler W, Kohlschütter A, et al. Language and cognition in children with metachromatic leukodystrophy: onset and natural course in a nationwide cohort. Orphanet journal of rare diseases. 2014;9(1):1-9. [View at Publisher] [DOI] [PMID] [PMCID] [Google Scholar]
12. JAbbeHDArI S, Rahimian E, Jafari N, Sanii S, Khayatzadehkakhki S, Biglari HN. The clinical features and diagnosis of metachromatic leukodystrophy: A case series of Iranian pediatric patients. Iranian journal of child neurology. 2015;9(3):57. [PubMed] [Google Scholar]
13. Golchin N, Hajjari M, Malamiri RA, Aminzadeh M, Mohammadi-Asl J. Identification of a novel mutation in ARSA gene in three patients of an Iranian family with metachromatic leukodystrophy disorder. Genetics and Molecular Biology. 2017;40:759-62. [View at Publisher] [DOI] [PMID] [PMCID] [Google Scholar]
14. Barboura I, Hadded S, Chebel S, Mansour RB, Chahed H, Gueddiche M-N, et al. Brain MRI and biological diagnosis in five Tunisians MLD patients. Diagnostic pathology. 2012;7(1):1-5. [View at Publisher] [DOI:] [PMID] [PMCID] [Google Scholar]
15. Groeschel S, Kehrer C, Engel C, í Dali C, Bley A, Steinfeld R, et al. Metachromatic leukodystrophy: natural course of cerebral MRI changes in relation to clinical course. Journal of inherited metabolic disease. 2011;34(5):1095-102. [View at Publisher] [DOI] [PMID] [Google Scholar]
Send email to the article author
| Rights and permissions | |
 |
This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License. |


This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International (CC BY-NC 4.0).